
- Disclaimer: This article is purely informative, and it does not contain information about how to actually carry out experimental procedures for the synthesis of LSD or any drug, which should NOT be attempted at home.
Lysergic acid diethylamide, commonly known as LSD, and colloquially called acid is a psychedelic drug which was first synthesized on November 16th, 1938 by a chemist called Albert Hofmann.
Do you want to known everything about the discovery and total synthesis of LSD? Keep reading!
LSD was discovered in Switzerland, but it was not until 1943 that the special properties of the compound were found. Today we do not focus on chemistry concepts but rather on a historical landmark. You might know a bit about LSD, but you also probably don’t know much more about its discovery and synthesis. That’s what we are going to fix in this article, it is a very interesting story and of course we will be covering a remarkable total synthesis!
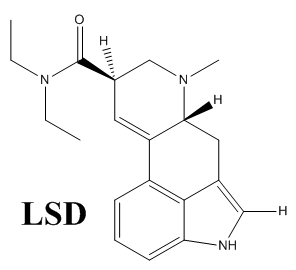
When it was discovered by Sandoz Laboratories, the purpose was using LSD as a respiratory and circulatory stimulant. It was found while analyzing organic compounds obtained from the ergot fungus and the medicinal plant squill.
LSD is well known for its psychological effects, which can give rise to closed- and open-eye visual hallucinations, alter the thinking process and the sense of time or, to sum up, induce abnormal psychic states. But as we have already seen, all of these properties were found no less than five years after its discovery, by the same guy who first synthetized it, Albert Hofmann. He was the first person to ingest and experiment the effects of the drug. The Telegraph newspaper placed him on the first position in a list of the 100 greatest living geniuses. The discovery of the psychoactive properties of LSD was a bit of a coincidence, since the Swiss chemistry accidentally absorbed a very small amount of the compound (the threshold dose is only about 20 micrograms) through his fingertips, finding these effects by himself. He also described how he was felling:
“…affected by a remarkable restlessness, combined with a slight dizziness. At home I lay down and sank into a not unpleasant intoxicated-like condition, characterized by an extremely stimulated imagination. In a dreamlike state, with eyes closed (I found the daylight to be unpleasantly glaring), I perceived an uninterrupted stream of fantastic pictures, extraordinary shapes with intense, kaleidoscopic play of colors. After about two hours this condition faded away”
The synthetic route that Hofmann used to prepare LSD is rather simple; the structure of the drug was very similar to the compounds extracted from the ergot fungus. He used ergotamine as starting material, so most of the structural work was already done by nature. We will focus in this article on the total synthesis of the drug, which means, a synthetic route that can be performed starting off from simple chemicals and reagents that are commercially available.
The newly-discovered physiological properties of ergot fungus also took Arthur Stoll attention, who also played a really important role on the early study of this family of compounds. He isolated and studied products such as ergotamine, ergonovine (the simplest one) and so on.
Growing Interest on the Synthesis of Lysergic Acid
The interest on the synthesis of lysergic acid rose from the discovery of all these compounds which had that part of the structure in common. The whole structure was not resolved and confirmed until 1949. However, it drew the attention of many organic and medical chemists anyway. Once the properties of LSD were found, this interest increased even more.
The first LSD synthesis was published on 1956, by one of the greatest (if not the most) organic chemists of all times, Robert Burns Woodward, born in Boston, Massachusetts. He is considered to be the best organic chemist of the 20th century, in terms of experimental and theoretical studies of chemical organic reactions. He also received the Nobel Prize in chemistry in 1965 for his synthesis of complex organic molecules. One of these molecules was lysergic acid. We will review his synthetic route as it deserves to be done. Also, we will cover the mechanism for each of the steps of this LSD synthetic route, in order to make it as instructive as possible for organic chemistry students. The original publication by Woodward can be found here.
The synthesis of lysergic acid presented an important problem: the high reactivity of its indole group. This heterocycle was considered so far incompatible with any long synthetic procedure. So, to avoid this problem, Woodward’s group decided to base most of the route on dihydroindole compounds (just like indole, but with 2 more hydrogens, and one double bond less), and transform it into the indole of LSD later one.The starting material of the whole route was β-carboxyethyldihydroindole, protected with a benzoyl group at the nitrogen.
The First Step: Ring C Formation
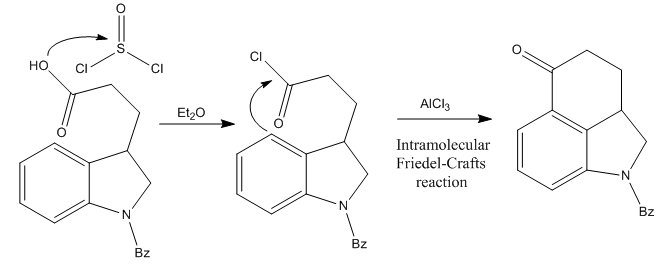
The initial compound was treated with thionyl chloride, converting it to the corresponding acid chloride. This makes the carbonyl group highly electrophilic. Then, the molecule undergoes an intramolecular Friedel-Crafts acylation reaction after the addition of aluminum chloride, assembling the ketone shown in the picture above.
Elaboration of the New Ring
The most problematic part of the synthetic route was the formation of ring D of the compound. Since they needed to add a substituent to the α-carbon to ketone carbonyl, a bromination was performed with some nasty molecular bromine in acidic media.
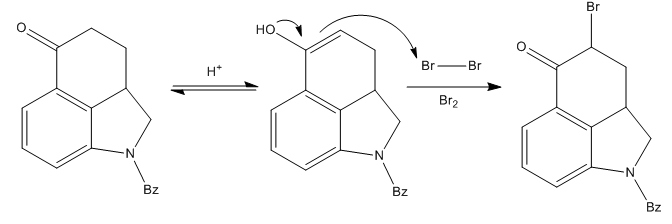
The desired brominated compound was obtained in a very good yield, but the first attempts to continue the synthesis from here failed. Many substitution reactions at the alkyl bromide failed.
However, after many unsuccessful attempts (and some successful but in rather poor yields), it was found that treating the brominated intermediate with methylaminoacetate ethylene ketal in a non-polar solvent, gave the desired alkylated intermediate in an excellent yield, which could be hydrolyzed using HCl to deprotect the acetal (releasing the ketone). At the very same time, the benzoyl group that protects the dihydroindole is also removed.
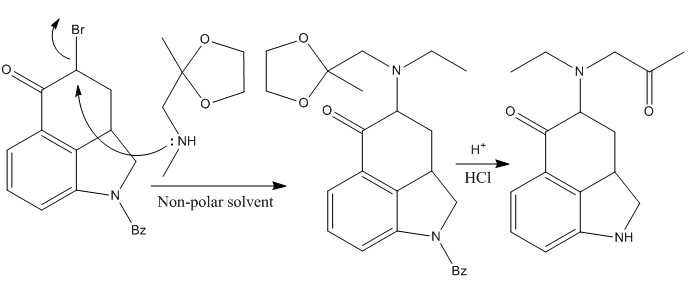
Finishing The Tetracyclic Core for the LSD Synthesis
The next step is the formation of the heterocyclic ring D, which was achieved effectively treating the last ketone intermediate with sodium methoxide in methanol.
The mechanism of this step is basically the formation of the kinetic enolate of the most accessible methyl ketone and then, nucleophilic addition of this enolate to the other ketone, closing the third and last ring of the molecule. This is immediately followed by an elimination reaction, giving rise to the corresponding α-β-unsaturated ketone.
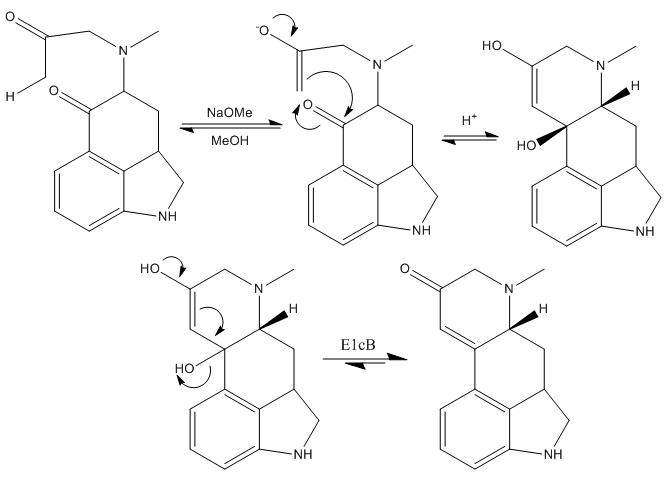
The treatment of this compound with sodium borohydride and sodium anhydride subsequentely reduces the ketone group to the alcohol, and protects the nitrogen of the dihydroindole. Next, they substituted the freshly introduced alcohol by a chloride. It was found that treating the alcohol with thionyl chloride in sulfur dioxide (liquid) gave the desired intermediate in good yield.
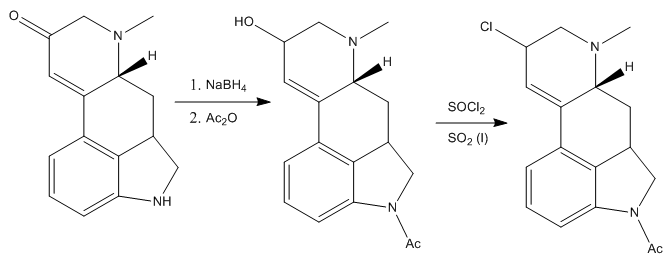
The obtained chlorinated intermediate was found to be very susceptible to hydrolysis to yield once again the alcohol, so the next reaction had to be performed fast and in special conditions: treating the compound with an excess sodium cyanide in anhydrous liquid hydrogen cyanide (pretty scary thing!). Anyway, Woodward’s group managed to make the reaction work and the resulting intermediate was treated with acidic methanol, to give the corresponding methyl ester. Also, the acetyl protecting group on the nitrogen was removed under the acidic conditions.
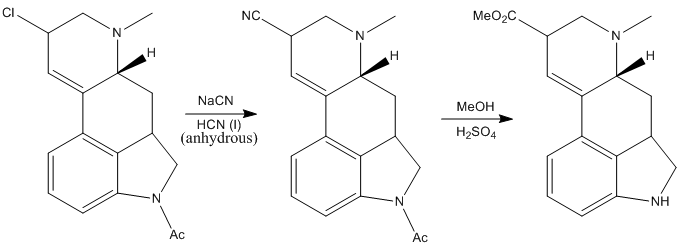
After this the product was hydrolized to give the corresponding carboxylic acid.
Now the work was almost done! The only remaining task to obtain lysergic acid was the formation of the oxidation of the dihydroindole to indole selectively. Some considerably obscure reaction conditions were employed, based on the use of Ni Raney and sodium arseante. This led to the desired indole (which is already lysergic acid), leaving untouched the rest of molecular functionalities.
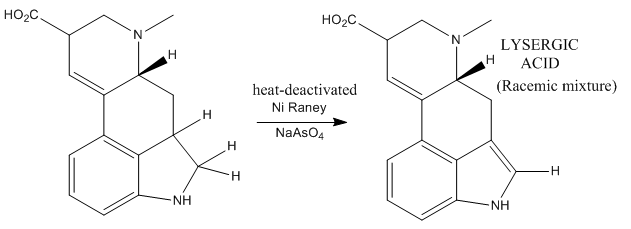
Lysergic acid was obtained as a racemic mixture which could be separated by chiral resolution.
Final Amidation of Lysergic Acid to Give LSD
However, this does not complete the synthesis of LSD. The last step is the formation of an amide bwith diethylamine.
The following are the reaction conditions used by Shulgin to obtain LSD from lysergic acid:
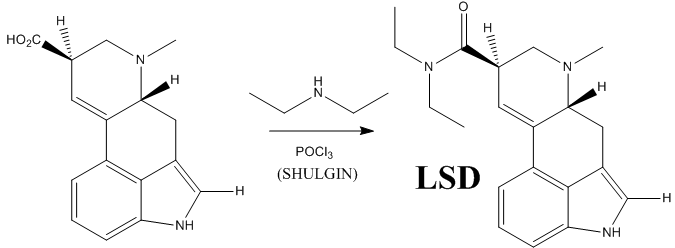
Alexander “Sasha” Shulgin was an American chemist author of the famous book PiHKAL: A Chemical Love Story (Phenylethylamines I Have Known And Loved), and its continuation, TiHKAL (Tryptamines I Have Known And Loved), where he makes a detailed explanation and analysis of how he discovered, synthesized and personally bioassayed a huge variety of drugs, all by himself (with the assistance of his wife, Aten Shulgin). He died less than a year ago, June 2, 2014 (aged 88), and with this last reaction I intended to make a small tribute to this great medicinal chemist, biochemist and psychopharmacologist.
You can PiHKAL books through Amazon, I promise they are worth a read!

“LSD,” -writes the chemist Alexander Shulgin– “is an unusually fragile molecule… As a salt, in water, cold, and free from air and light exposure, it is stable indefinitely.”
But of course, this is not the end of the story… Organic chemistry and synthetic techniques have advanced A LOT from those years to present, and way better methods have been published to prepare lysergic acid diethylamide from scratch in a more efficient and easier way.
Modern LSD Synthesis Routes
A very recent route for the total synthesis of LSD is that published by Tohru Fukuyama et al. from the Graduate School of Pharmaceutical Sciences, University of Tokyo, in 2013.
This LSD synthesis is based on the Evans aldol reaction, which allows a stereoselective construction of the needed chiral center followed by a sequential process, which includes a metathesis reaction that produces the ring-closure and finally a Heck reaction which finishes the construction of the two rings.
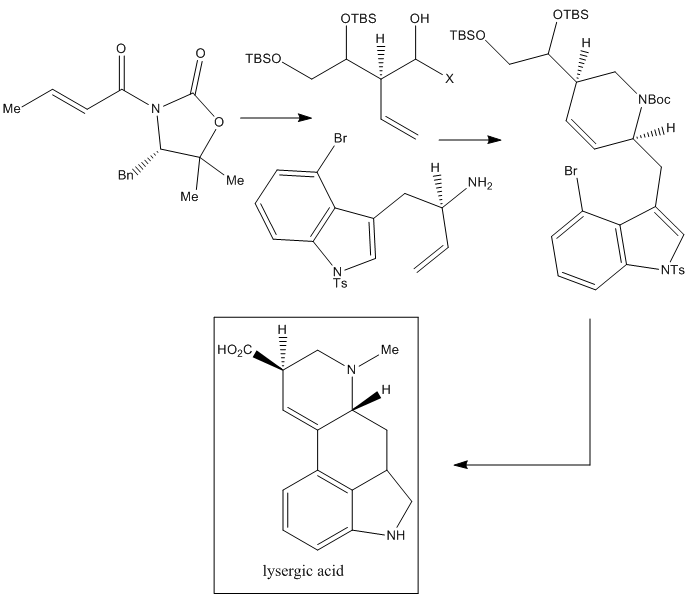
To finish this article, I would like to say that this is just an example of the discovery, isolation, preparation and development of a new kind of drug, and many others have been discovered over the years, which have saved and improved (and of course, still do) the life of humanity. The fact that LSD can be used as a recreational drug is not the topic of this review.
I enjoy organic and medicinal chemistry a lot (it is actually a part of my life, what I studied and what I work on every day), and I love writing this kind of articles. Please, do share your thoughts, criticism, or suggestions in the comments, it will be really appreciated. Feedback encourages us to keep working on Chemistry Hall!
Organic chemistry can explain a great deal of things. If you want to take a look at another real-world chemistry story, I’d recommend you to dive into why urine smells bad after eating asparagus, as we covered in another post.
Also, if you have any suggestion or idea for future posts, it will be strongly valued too!
Very good read! Thank u
Thank you for the feedback, it is reall important to us!
What’s interesting is that lysergic acid is also the natural precursor of all therapeutic ergot alkaloids. The current industrial method of lysergic acid production is based on decaying matter which fuel fermentations by Claviceps cultures. After purification, pharmaceutical companies end up with forty thousand pounds of lysergic acid every year. A few years ago Harvard’s Department of Systems Biology were in the process of sneaking a section of Claviceps genome into baker’s yeast to get it to produce lysergic acid. I haven’t followed up on the research to see where it’s at now.
Sounds really interesting, will check it out whenever I have some free time, thanks for sharing and for the opinion!
Another interesting note. The Lysergic acid production with Clalviceps is the most difficult part to accomplish. Even before the synthesis of a lifetime! if anyone can show me proof ever being successful with lysergic acid production I would be shocked.
Excellent article here. Another good resource that covers a subject like this is The Chemistry of Mind-Altering Drugs – Daniel M. Perrine. Please keep up the great work.
Will do! Thanks to people like you that are telling me that enjoy the reads! Regards
You have the structure for LSD wrong, you’re showing an Ac on the indole nitrogen instead of H. You’re showing the structure of ALD-52!
Nicely spotted! Thanks for the correction, I’ve already fixed it
The structure shown as LSD is not LSD but ALD-52. LSD is not acetylated at the indole nitrogen.
Thanks for correcting it, it’s already fixed. Seems like the deprotection step didn’t work in a very high yield for me haha
I am amazed by the understanding of this advanced chemistry, and inspired by the ideas behind these discoveries which confirm older theories, as well as spawning even more ideas. You all rock!
Very respectfully – Omar
The first total synthesis of lysergic acid was NOT by R.B. Woodward. It was by Ed Kornfeld, at Eli Lilly. Ed had moved to Eli Lilly after his postdoc and they hired Woodward as a consultant. It is a very common mistake to attribute the total synthesis to Woodward because he was such a great chemist so everyone just assumes he did it. Also, Sasha Shulgin never had a license that allowed him to make LSD in his lab. He could only make unscheduled molecules, and LSD was a schedule I substance. The method he describes was the one published by Fatima Johnson et al. in J. Med. Chem. 16:532-537 (1973).
Good job, Dave! Facts are like sheep—they tend to wander and need a good sheepdog to nip their heels!
Dan Perrine
Currently, part of the Internet is full of eyeglasses retailers of numerous styles and differing levels of popularity.
The constant moving and moving of one’s eyeglasses can continually annoy each you as well
as other those who discover you.
I really don’t know anything about synthesizing different chemicals so I’m going to aplogize in advanced if I say something stupid. Some of the chemicals (like cyanide) they use during the process of making lsd sound like they can have dangerous health effects. Do those chemicals still effect what the drugs effects on your body? From what I know the potential neggative effects of lsd are psychological and not physical. Do these chemicals used add potential physical damage if you take lsd? Or are chemicals like this used often during the sythesis of different chemical and are changed to a point where it negates the dangers?
Hey,
A very large deal of the drugs that are produced and used in tons every day to treat diseases are synthesized using chemicals or reactants that would be quite poisonous or dangerous, but the drugs itselves are quite safe in the appropriate dosage. When you make a chemical react with another (like when you are building a molecule) the chemical properties/interaction with our bodies of the product you obtain will be completely different to those of the starting materials (if they were to be the same or very similar it would just be a coincidence).
In the case that you mention the properties of inorganic hydrogen cyanide (reagent) are completely different from the ones of an organic nitrile (product).
In industry (or in academic labs), after every step of the synthesis, the products are put through a purification step which removes every dangerous/unwanted chemical and leaves only the final product. This is especially important in pharma or food industry, where there is a very strict control of impurities present on their products.
One of the problems of “recreational drugs” is exactly this one: Many “street-made” drugs may have not been purified properly, so the drug is not the problem itself, but its level of impurities generated along the synthetic process.
Coll, possession of more than 5 doses of LSD was to be considered smaller than large for the purposes of the Criminal Code and was to be treated as a misdemeanor subject to a fine equal to a parking ticket.
Just stumbled across this in a google search. Thanks.
My wife and I spent a long afternoon with Albert Hofmann (and Anita) in his home, back in 2003. He told us we were the first folks, outside his family and old friends, invited to his home in over 20 years. He was 97 then, a gracious host and great storyteller – the one of his friendship with Huxley was especially enjoyable, poignant. He invited us to his 100th birthday party/LSD Symposium in Basel, and we went. It was a great event.
Others I’ve known had far more history with Albert Hofmann. But that one afternoon at his Swiss home, where he got me tipsy on his homemade plum schnapps (and I’m a teetotaler – but it was Albert Hofmann…), and discussed the problems, and wonders, associated with being the father of LSD, is a priceless memory for me and my wife.
Thanks again for the informative article.
Residential roofing will get families out of the chilly.
Totally off topic, in the early 70’s I had the opportunity to act as Albert Hoffman’s chauffer. One afternoon we went to Port Townsend Washington and stopped at a tavern for a pitcher of beer. While we were drinking our brew a gentleman with hair down to his waist was twirling around playing the flute. The expression on Mr. Hoffman’s face was one of amused resignation. True story!
That is absolutely awesome! You must be full of nice stories that people may want to read. Feel free to contact me at infochemistryhall@gmail.com if you ever want to share them on this blog 🙂
David Jewell, I imagine that you
chauffeured Albert to the 1976 Hallucinogenic Mushrooms Conference in Port Townshend, Washington? The one put on by Jeremy Bigwood, & Jonathan Ott?
A historic moment in time ghat you were part of. Thanks for sharing.
I intend no offense, but you should consider using a proof reader. This is an interesting story, one I am familiar with both historically and chemically, but it is littered with grammatical errors which could easily be corrected by someone having the appropriate expertise.
You are right. I’m not a native English speaker, and at the time I wrote this article (almost 6 yeas ago now!), I wasn’t exactly fluent. I appreciate your advice and consider doing a revision of my older posts when I get the chance.
Very great read. Thank you for your hard work!
I would love to have a few squares of window pane right now!
Wouldn’t we all. Psilocybin cubensis is easier to obtain/ produce. Shorter duration, but just as intense. Funny, ergot and cubensis both love rye grain.